
Hong Kong J Psychiatry 2002;12(3):12-18
REVIEW ARTICLE
Xin Duan, GY Ma
Dr Xin Duan, Department of Psychiatry, Shantou University Medical College, Shantou, China
Dr GY Ma, Professor of Psychiatry and Forensic Medicine, Deputy Chief of Mental Health Expert Group of Guangdong Province, Department of Psychiatry, Shantou University Medical College, Shantou, China
Address for correspondence: Dr Xin Duan, Department of Psychiatry, Shantou University Medical College, 22 Xin Ling Road, Shantou
515031, Guang Dong Province, China. E-mail: g_xduan@yahoo.com.cn
Submitted: 15 September 2001; Accepted: 14 January 2002
Abstract
Alzheimer’s disease can either occur sporadically or be caused by inheritance of a gene mutation accompanying progressive loss of memory and general decline in cognition. The pathology of Alzheimer’s disease is complex, but there are 2 hallmarks: the neuritic plaque consisting of b-amyloid derived from b-amyloid precursor proteins and the neurofibrillary tangles consisting of abnormally phosphorylated tau protein. Transgenic mice are used to reproduce these 2 hallmarks since they can reproduce many features of the disease. This review provides an overview of transgenic mouse models including familial Alzheimer’s disease and non- hereditary Alzheimer’s disease as well as emerging insights relevant to the pathogenesis and potential treatment strategies.
Key words: Alzheimer disease, Animal models, Therapy, Transgenic mice
Introduction
Alzheimer’s disease (AD) is the most prevalent type of de- mentia. It occurs either sporadically or is caused by inheritance of a gene mutation accompanying progressive loss of memory and global cognitive decline. The pathology of AD is complex, but there are 2 hallmarks that occur abundantly in most cases. The neuritic plaque is a largely extracellular lesion consisting of b-amyloid (Ab) released from cleaved Ab precursor proteins (APPs) by a-, b- and g-secretase and the intracellular neurofibrillary tangles (NFTs) consist largely of abnormally phosphorylated tau protein. Neuronal and synaptic loss with reactive gliosis also occur.
A good animal model will help considerably both for understanding the relationships between various aspects of pathology and for testing therapies based upon these relationships.1
Hereditary Alzheimer’s Disease Transgenic Animal Model
Alzheimer’s Disease Genetics
Some cases of early onset AD are familial autosomal dominant (FAD) disorders caused by mutations in APP, PS1 and PS2.2 In the late onset forms, there are no specific gene mutations associated with FAD inheritance, although specific alleles of apolipoprotein E and a2 macroglobulin (A2M) are reported to be associated with increased risk for AD (Table 1).3
Transgenic mice that express disease-causing genes reproduce many features of the disease. These models are prov- ing to be useful in investigations of the nature of biochemical alterations in neural tissue, the character and evolution of pathologies, and the pathogenic mechanisms.
b -Amyloid Precursor Protein Mutations
The APP gene, the first AD susceptibility gene to be iden- tified, encodes a transmembrane protein that contains 770 amino acids in its longest isoform. Mutational studies of the APP gene have identified a Glu693Gln missense mu- tation of the APP gene,4 and several other missense mutations in exons 16 and 17 of the APP gene. Although a number of APP mutations have been identified, most are probably not disease causing. However, the missense mutations at codon 670/671 (Swedish mutation), at codon 692 (Flemish mutation), at codon 716, and at codon 717 are thought to be pathogenic.
The first successful transgenic mouse model with Ab amyloid deposition was made by Games et al.5 These researchers took an unusual construct that included full- length human APP complementary DNA with the APP717Val(V)®Phe(F) mutation under the control of the platelet-derived growth factor promoter, which targets expression preferentially to neurones of the cortex, hippo- campus, hypothalamus, and cerebellum of transgenic animals.6 The pathology of the mice was as striking as in patients with AD, and included extracellular Ab deposition, dystrophic component, gliosis, and loss of synaptic density with regional specificity resembling that of AD, although NFTs were not evident.

Hsiao et al used the hamster prion protein promoter to overexpress the human APP 695 containing Lys670®Asn, Met671®Len mutation (Swedish mutation).7 These transgenic mice had normal learning and memory in spatial reference and alternation tasks at the age of 3 months, but showed impairment by the age of 9 to 10 months. This impair- ment was correlated with a marked increase in the amounts of Ab and was accompanied by numerous amyloid plaques and Ab deposits.
The transgenic mice with robust behavioural and patho- logical features resembling those found in AD offer an appropriate tool for exploring the pathophysiology and neurobiology of this disease.7 Both studies support the amyloid cascade hypothesis of AD pathogenesis, which states that production and deposition of Ab in the form of fibrils leads to neuronal cell death and eventually to clinical presentation and progression of AD.8
Genetic strategies have also been used to gain insight into mechanisms of the disease. A controversial question still exists as to whether Ab-induced neurotoxicity requires deposition of aggregated Ab into plaques.9-12 Hsiao et al postulated that the neurotoxic effect of Ab is independent of plaque formation.7
Transgenic mice with overexpression of human APP with V717F mutation and addition of the Swedish FAD mutation to the APP gene were studied. These researchers observed that the density of presynaptic terminals and neurones had decreased well before the FAD (V717F)- mutant human APP transgenic mice developed amyloid plaques. Electrophysiological recordings from the hippo- campus revealed prominent deficits in synaptic transmission, which also preceded amyloid deposition by several months. Increased Ab production in the context of the decreased overall APP expression, achieved by the double transgenic mice (DTG mice), further increased synaptic transmission deficits in the young mice without plaques.13 The result supports the new hypothesis that the soluble Ab forms (protofibrils and small oligomers) could affect neurones, but escape detection by measurement of solid amyloid.14
PS Mutations
Genetic linkage studies mapped a locus associated with a aggressive early-onset AD to a series of polymorphic markers located on chromosome 14q24.3. The actual disease gene (PS1) was subsequently isolated using a cloning strategy15 and a homologue (PS2) was then mapped to chromosome 1q42.1.16,17 To date, more than 70 different mutations have been discovered in the PS1 gene, the majority of these mutations are missense mutations, giving rise to the sub- stitution of a single amino acid. There are 6 different mutations in the PS2 gene. In contrast to the high frequency of PS1 mutations, screening of large data sets revealed that PS2 mutations are probably rare.
To explore the functions of PS1 and PS2 genes, the best approach is a gene knockout strategy. PS1 deficient mice (PS1-/- mice) die late in embryogenesis without evidence of AD. It has been proposed that this phenotype is a result of disturbed Notch signaling.18,19 PS1 is also involved in normal APP processing in neuronal cultures derived from PS1- deficient mouse embryos. PS1 mice have a defect in APP processing manifested by the failure of g-secretase cleavage and the accumulation of c-terminal stubs of APP following a- and b-secretase cleavage. This indicated that PS1 is involved in g-secretase activity.20 This defect in APP pro- cessing is completely reversed by both wild-type and mutant PS1 genes. However, in contrast to PS1-/- mice, PS2-/- mice show no major developmental or APP processing defects.21
Accumulating studies showed that mutant PS1 and PS2 genes increase the production of amyloidogenic Ab42 and Ab43 peptides.22-25 Transgenic mice that express either wild type PS1 or A246E PS1(Ala246®Glu mutation) were generated and mated with Mo/Hu-APPswe mice.26
Similarly, Holcomb et al mated PS1 transgenic mice with APPswe transgenic mice (line Tg2576).27 Both studies demonstrated that mice coexpressing mutant PS1 with APPswe developed Ab deposits much earlier than age- matched animals that express APPswe alone, mutant PS1 alone, or wild-type PS1 with APPswe. In these transgenic models of Ab amyloidgenesis, NFTs were not described. Moreover, neuronal loss is only observed in mice with a high amyloid burden.
b-Amyloid Precursor Protein Proteases
Although different subsets of AD phenotypic traits are reproduced in these transgenic models, none of them repro- duce all the features typical of AD. Nevertheless, available studies have provided evidence to support the view that Ab42 formation is an early and critical pathogenic event.28
Ab occurs in 2 predominant forms (Ab40 and Ab42) and is generated from the amyloid precursor protein by protein-cleaving (proteolytic) enzymes called b-secretase and g-secretase.29 AD-causing mutations in APP flank the protease cleavage site in APP and facilitate its cleavage. Those near the b-secretase cleavage site augment b-site proteolysis, leading to elevation of both Ab40 and Ab42; whereas mutations near the g-site specifically increase production of Ab42. AD-causing mutations of presenilins modulate g-secretase activity to enhance production of Ab42 (Figure 1)
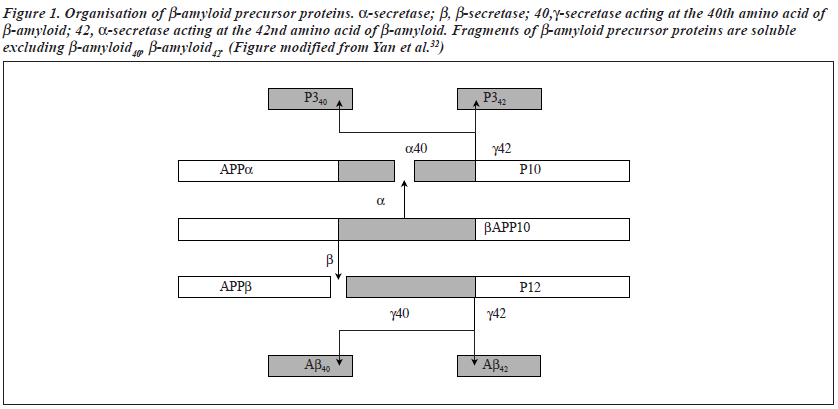
b-secretase and g-secretase were identified in 1999 and in 2000, respectively.30-34 b-secretase (beta site APP cleaving enzyme or BACE) is a new transmembrane aspartic protease. The gene for b-secretase (or BACE) is located on chromosome 11.35 g-Secretase is also a new transmembrane aspartyl protease. Accumulating evidence suggests that presenilins may be the catalytic component of g-secretase, and may even be g-secretase.36 Li et al showed that PS1 (and PS2) contain the active site of g-secretase, and transition- state-specific g-secretase inhibitors specifically bind to the presenilins.37 Li et al further showed that presenilin 1 is first synthesised as a zymogen (an inactive precursor protease), which does not bind to the inhibitor.37
b-secretase and g-secretase could be potential pharmaco- logical targets. Blocking the 2 responsible proteases would be a reasonable approach for treatment interventions during the development of AD pathologies. Development of any medication requires validated animal models and well-defined molecular targets.38
The above results could, perhaps, accelerate the develop- ment of a drug to stop or even reverse the neurodegenerative process. Intense efforts should be directed toward the development of clinically useful secretase inhibitors. Several challenges remain. The main problem will be the delivery of such drugs to targets in the neurones through several formidable penetration hurdles, such as the blood-brain barrier and lipid bilayer of the subcellular space.
Another important problem is that these drugs should be highly selective: interference with other intracellular proteases and critical signalling pathways must be minimised. Further suitable experimental animal models will be needed to facilitate future studies.
Apolipoprotein E Alleles
Apolipoprotein E (ApoE), the major serum protein involved in cholesterol storage, transport, and metabolism, is poly- morphic and encoded by 3 alleles (apoE2, apoE3, and apoE4), which are located on chromosome 19q12-q13.39,40
ApoE3, the most common variant, reflects the presence of a cysteine at codon 112 and arginine at codon 158. ApoE4 reflects substitution of arginine for cysteine at codon 112. ApoE2 contains cysteine at codons 112 and 158. Numerous studies and data have confirmed the association between apoE4 and AD. The presence of 1 or 2 E4 alleles is asso- ciated with earlier onset of disease and enhanced amyloid burden in the brain.41,42 On the other hand, the E2 allele may be protective in this aspect.43
Transgenic mice were created using human apolipo- protein E2, E3, E4 gene fragments driven by the human glial fibrillary acidic protein (GFAP) promoter. Smith et al utilised selected lines by breeding with apoE-deficient mice yielding mice which express the human transgenic apoE isoforms in the absence of endogenous apoE.44 Aged apoE4 transgenic mouse brain fails to demonstrate any evidence of senile plaques.
However, APPV717F transgenic mice on a wild-type murine apoE show profuse amyloid deposition. In contrast, when the same transgene is expressed in an apoE-/- back- ground, there is only sparse, diffuse Ab deposits.
The level of total Ab and Ab42 in the hippocampus of APPV717F: apoE-/- mice as measured by enzyme-linked immunosorbent assay is significantly less than that found in APPV717F: apoE+/+ mice. The ratio of total Ab42/Ab, however, is not modified by the absence of apoE in this model. The effect of apoE on fibrillogenesis in vivo does not appear to act through alteration of Ab42-to-total Ab ratio.
This finding and the fact that APP levels do not differ between animals with and without apoE suggest that apoE is influencing Ab metabolism, structure, and/or clearance after being processed and released from APP.45 Thus, it appears that the apoE allele type is not causative, but rather modulates the disease process.
To determine whether human apoE is also required for the development of fibrillar Ab deposition and neuritic plaques in APPV717F transgenic mice, Holtzman et al bred APPV717F+/+: apoE-/- mice to apoE3 and apoE4 transgenic mice expressing human apoE isoforms under the control of the astrocyte-specific GFAP promoter.46 Ab deposition occurs in the presence of both apoE3 and apoE4. There is a significant isoform-specific difference in the amount of deposition, with greater Ab deposition in apoE4 as compared with apoE3 expressing mice. These data show that apoE is required for amyloid formation with the FAD- mutant APP background and apoE4 has a greater effect on Ab deposition than apoE3.
To further explore whether apoE4 simply does not function as well as apoE3 or whether it exerts detrimental effects that may interfere with the neuroprotective function of apoE3, Buttini et al analysed transgenic apoE knock-out mice that express apoE3 or apoE4 or both in the brain.47
Apolipoprotein E3/E4 bigenic mice are as susceptible to neurodegeneration (by intraperitoneal injection of the glutamate receptor agonist, kainic acid) as apoE4 singly- transgenic mice (E4/0). Thus, apoE4 acts as a dominant negative factor that interferes with the beneficial function of apoE3. At 8 months of age, neurodegeneration is more severe in homozygous (E4/E4) than in hemizygous apoE4 (E4/0) mice.
This result indicates that the detrimental effects of apoE4 are dose-dependent. The exact mechanisms under- lying the detrimental apoE4 activity remain to be deter- mined. These experiments, consistent with other studies, strongly support that apoE4 acts as a genetic risk modifier for AD.
Alzheimer’s Disease Vaccine: a Promising Therapy
These transgenic animal models can offer a viable means to test whether compounds can produce beneficial effects in an animal model prior to advancing such a drug into human trials. Schenk et al first reported that Alzheimer’s- like pathology in mice could be halted by vaccination with solutions of Ab peptide.48 These researchers used PDAPP transgenic mice, which overexpresses human mutant APPV717F. The transgenic animals were immunised with Ab42 either before the onset of AD-type neuropathology (at the age of 6 weeks) or at an old age (11 months), when b-amyloid deposition and other subsequent neuropatho- logical changes were well established.
Some effects were noted — plaques were largely prevented from forming, and some of the pre-existing plaques in older mice even dissolved. Outcomes of Ab- plaque burden, neuritic dystrophy, and liosis were signifi- cantly improved by Ab42 treatment in both young and old animals.
In addition, the mechanism resulting in plaque reduction does not seem to produce any obvious signs of damage to the neuropil of Ab42-immunised animals. Histological examination of several organs, including the brain and kidney, revealed no signs of immune-mediated complica- tions, despite the high levels of human APP expressed in these tissues and the significant antibody titre to endogenous mouse Ab peptide. These researchers observed that Ab42 immunisation resulted in the generation of anti-Ab antibodies and that Ab-immunoreactive monocytic/microglial cells appeared in the regions of remaining plaques.
They postulated that the possible mechanism of action is that anti-Ab antibodies facilitate clearance of b-amyloid either before deposition or after plaque formation by triggering monocytic/microglial cells to clear b-amyloid using signals mediated by Fc receptors.48 This view seems to be in harmony with either the soluble Ab hypothesis or the amy- loid cascade hypothesis. The antibodies derived from immunoreaction of soluble Ab may become useful tools for further identifying toxic peptides, and provide insights in future studies.
It remains unknown whether the vaccine could also stave off the cognitive losses that make Alzheimer’s disease so devastating and whether the vaccine is safe enough to use to treat AD. Morgan et al tested the effects of Ab vaccination in a different transgenic model (APPK670N, M671L + PS1M146L) in which the mice developed learning deficits as amyloid accumulated.49
The experimental vaccine was prepared from human Ab1-42 (Bachen) and produced a sustained high titre level in the DTG mice. The control vaccine was prepared from keyhole limpet hemocyanin (KLH). The researchers tested mouse behaviour (‘episodic-like’ memory) in a radial arm water maze working memory task. The results showed that Ab vaccination protects transgenic mice from developing memory deficits compared with KLH-immunised (control) transgenic mice. This vaccination-associated protection occurs in the presence of reduced, but still substantial, Ab deposits. To address this finding, Morgan et al hypothesised that antibodies produced by vaccination either neutralise Ab in a restricted neuronal compartment or deplete a non- deposited form of Ab (for example, a soluble form).49
Another laboratory found similar results that immunisation against Ab (in b-pleated-sheet conformation) not only reduces plaque formation, but also ameliorates cognitive deficits in the TgCRND8 murine model of Alzheimer’s disease that expresses a mutant (K670N/M671L + V717F) human APP695 transgene.50 These results strongly support a causal link between a high level of Ab and cognitive decline.51
Two groups suggest that either a small or a selective reduction in b-amyloid deposition may be sufficient to protect against dementia52 and both studies offer a promise regarding the safety and efficacy of vaccination with Ab peptide vaccines. Potential applications in human subjects will require knowledge of the therapeutic mechanism and the specific antigens being targeted. Further intensive investiga- tion will be needed to identify the best approach for safe and effective vaccine development.53
Tau and Amyloid Plaques
Neurofibrillary tangles (NFTs) composed of the microtuble- associated protein tau are prominent in AD, Pick’s disease, and other neurodegenerative diseases. Mutations in the gene encoding tau protein cause frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). This demonstrates that there is a link between tau dysfunction and neurodegeneration. At least 11 missense mutations and a 3-base pair deletion (DeltaK 280) have been identified in exons 9 to 13. Additionally, 5 splice site mutations have been found in intron 10.54 The most common mutation P301L (proline®leucine) derives from a C-to-T change in exon 10 on chromosome 17q21-22.55,56 However, no AD- causing tau mutations have been identified at present.
Gotz et al expressed human tau in P301L transgenic mice by using the neuron-specific mouse Thy1.2 promoter.57 High expression of human P301L tau is obtained in cortical and hippocampal neurones in the mice. Accumulated tau is hyperphosphorylated and translocated from axonal to somatodendritic compartments and is accompanied by astrocytosis and neuronal apoptosis. As expected, P301L tau forms abnormal filaments. Neurofibrillary tangles occur in the cortex, brainstem, and spinal cord. The results showed that expression of the P301L mutation in mice causes neu- ronal lesions that are similar to those seen in human tauopathies. The conclusion is similar to that of Lewis.58
There is a debate as to whether b-amyloid or tau is the primary cause of AD pathology and what is the patho- physiological relationship between them. Hardy et al proposed that the peptide Ab42 is central to the aetiology of AD and tau is produced either indirectly, by Ab42, or directly, in some form of frontotemporal dementia by mutations in tau itself.59 Gotz et al injected synthetic Ab42 fibrils into the somatosensory cortex and hippocampus of 5- to 6-month-old P301L tau transgenic mice.60 They observed 5-fold more NFTs in Ab42-injected animals than in animals injected with a control peptide.
NFT formation was found in the amygdala, remote from the injection site in hippocampus. Meanwhile Lewis et al crossed Tg2576 transgenic mice expressing the APPsw mutation (Lys670Asn, Met671Lew) with JNPL3 transgenic mice expressing mutant P301L 4-repeat tau and compared the pathology of the crossed mice with each of their paren- tal lines.61 The resulting double mutant (tau/APP) progeny and the Tg2576 parental strain develops Ab deposits at the same age; however, relative to JNPL3 mice, the double mutants exhibit NFT pathology that is substantially enhanced in the limbic system and olfactory cortex. Both studies confirmed that Ab42 fibrils can significantly accelerate NFT formation in transgenic mice and revealed interaction pathologies in AD between APP or Ab and tau.
Transgenic models reproducing the 2 pathological hallmarks of AD will help in the search for more effective therapies. The challenge for Ab vaccination for AD treatment will be whether it can prevent NFT formation in animal models. If it is effective in retarding the formation of both amyloid plaques and NFTs in the transgenic models, the hypothesis of b-amyloid as a causative pathogenic factor in AD could be better established. AD will no longer be an unbeatable disease and finding therapeutic interventions will no longer be an impossible dream.
Non-hereditary Alzheimer’s Disease Transgenic Animal Models
Non-hereditary AD transgenic animal models emerged during the past year. Capsoni et al succeeded in making anti- nerve growth factor transgenic mice.62 Nerve growth factor (NGF) is widely distributed in the basal forebrain choliner- gic neurones (BFCNs) and in regions of the central nervous system innervated by the magnocellular BFCNs. NGF promotes the differentiation of BFCNs,63 ameliorates lesion- induced abnormalities in these cells,64 and reverses atrophy of BFCNs65 and spatial memory impairments in aged rats.
Studies propose that NGF might be used as a potential therapeutic agent to prevent the degeneration of BFCNs in AD patients. However, the lack of suitable animal models in which the activity of NGF is chronically blocked in the adult central nervous system has not allowed proof of whether a reduced level and/or efficacy of NGF signalling may play a role in the pathogenesis of AD. The heterozy- gous NGF knockout mice (ngf+/-) made by Chen et al showed shrinkage of basal forebrain and hippocampal neurones and a 20% reduction of ChAT-positive BFCNs associated with behavioural deficits.66 However, the authors did not report any sign of AD-like neuropathology.
Capsoni et al used the neuroantibody technique to pro- duce transgenic mice expressing a neutralising mAb (mAb aD11) directed against NGF, in which the levels of anti- bodies (a neutralising anti-NGF recombinant antibody) are 3 orders of magnitude higher in adult than in newborn mice.62 They reported that aged anti-NGF mice show massive and widespread neuronal loss, amyloid deposits, and extensive neurofibrillary pathology demonstrated with antitangle and antiphosphorylated tau antibody.
Moreover, these mice exhibit a severe cholinergic defi- cit in the basal forebrain and a behavioural impairment in retention and transfer of spatial memory tasks. 62,67 Also, the progression of the neurodegeneration observed in anti- NGF mice showed a positive correlation with the severity of pathology, with the transentorhinal region showing the first signs of neurofibrillary lesions. Thus, the group con- sidered that this phenotype is a comprehensive transgenic model for AD research. Recent studies have shown the link between NGF and amyloid plaque, but the interaction mechanism is not clear.68,69
NGF may be a potential therapeutic agent for the treatment of AD, but the problems of CNS delivery and side effects (particularly pain) still need to be resolved. Pharmacological approaches to enhance production of NGF in the CNS may also be useful in the treatment of AD.70
Conclusion
In brief, much progress has been made towards under- standing the aetiologies of AD in the past decade. Genetic models now available offer hope in the field of neuroscience. Knockout animals offer important clues to normal function. Transgenic animals not only allow investigation of the patho- logical role of mutant genes, but can also provide important models for drug development.71
Experimental data from AD transgenic mice models that reproduce both amyloid plaques and NFTs provide further support for the hypothesis that b-amyloid may be a caus- ative pathogenic factor. The question of whether an increase in the level of Ab or the ensuing plaque formation is the causal factor in AD remains controversial. With the appearance of the soluble Ab hypothesis, transgenic animals with CNS deficits were once considered poor models of AD pathogenesis if they lacked amyloid deposits. At present, it appears that such animals provide good models for pathogenesis by non-fibrillar Ab. These animal models rein- force the weak correlation between amyloid and disease in humans.15 Strategies that modulate the production, deposi- tion, or toxicity of Ab might be considered reasonable thera- peutic approaches. Currently, reduction of Ab levels is widely thought to be the most likely to succeed and means such as Ab vaccination and agents that inhibit Ab aggregation hold great promise.
References
- Duff K, Hardy J. Alzheimer’s disease: mouse model made. Nature 1995;373:476-477.
- Price DL, Sisodia SS. Mutant genes in familial Alzheimer’s disease and transgenic models. Annu Rev Neurosci 1998;21:479-505.
- Mayeux R, Saunders AM, Shea S, et al. Utility of the apolipoprotein E genotype in the diagnosis of Alzheimer’s disease. Alzheimer’s Disease Centers Consortium on Apolipoprotein E and Alzheimer’s Disease. N Engl J Med 1998;338:506-511.
- Levy E, Carman MD, Fernandez-Madrid IJ, et al. Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 1990;248(4959):1124-1126.
- Games D, Adams D, Alessandrini R, et al. Alzheimer-type neuropa- thology in transgenic mice overexpressing V717F beta-amyloid pre- cursor protein. Nature 1995;373:523-527.
- Sasahara M, Fries JW, Raines EW, et al. PDGF b-chain in neurones of the central nervous system, posterior pituitary, and in a transgenic model. Cell 1991;64:217-227.
- Hsiao K, Chapman P, Nilsa S, et al. Correlative memory deficits A b elevation, and amyloid plaques in transgenic mice. Science 1996;274: 99-102.
- Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature 1999;399(6738 Suppl):A23-31.
- Cummings BJ, Pike CJ, Shankle R, Cotman CW. Beta-amyloid depo- sition and other measures of neuropathology predict cognitive status in Alzheimer’s disease. Neurobiol Aging 1996;17:921-933.
- Bartoo GT, Nochlin D, Chang D, Kim Y, Sumi SM. The mean A beta load in the hippocampus correlates with duration and severity of de- mentia in subgroups of Alzheimer disease. J Neuropathol Exp Neurol 1997;56:531-540.
- Terry RD. The pathogenesis of Alzheimer disease: an alternative to the amyloid hypothesis. J Neuropathol Exp Neurol 1996;55:1023-1025.
- Gomez-Isla T, Hollister R, West H, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol 1997;41:17-24.
- Hsia AY, Masliah E, Mclonlogue L, et al. Plaque-independent disrup- tion of neural circuits in Alzheimer disease mouse models. Proc Natl Acad Sci USA 1999;96:3228-3233.
- Klein WL, Krafft GA, Finch CE. Targeting small Ab oligomers: the solution to an Alzheimer’s disease conundrum? Trends in neuroscience 2001;24:219-224.
- Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995;375(6534):754-760.
- Levy-Lahad E, Wijsman EM, Nemens E, et al. A familial Alzheimer’s disease locus on chromosome 1. Science 1995;269(5226):970-973.
- Rogaev EI, Sherrington R, Rogaeva EA, et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 1995;376(6543): 775-778.
- Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell 1997;89: 629-639.
- Wong PC, Zheng H, Chen H, et al. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature 1997;387(6630): 288-292.
- De-Strooper B, Saftig P, Craessaerts K, et al. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998; 391(6665):387-390.
- Herreman A, Hartmann D, Annaert W, et al. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc Natl Acad Sci USA 1999;96: 11872-11877.
- Duff K, Eckman C, Zehr C, et al. Increased amyloid-b 42(43) in brains of mice expressing mutant presenilin 1. Nature 1996;383:710-713.
- Borchelt DR, Thinakaran G, Eckman CB, et al. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron 1996;17:1005-1013.
- Martins RN, Turner BA, Carroll RT, et al. High levels of amyloid-beta protein from S182 (Glu246) familial Alzheimer’s cells. Neuroreport 1995;7:217-220.
- Scheuner D, Eckman C, Jensen M, et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med 1996;2:864-870.
- Borchelt DR, Ratovitski T, van-Lare J, et al. Accelerated amyloid depo- sition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 1997;19:939-945.
- Holcomb L, Gordon MN, McGowan E, et al. Accelerated Alzheimer- type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 1998;4:97-100.
- Price DL, Sisodia SS, Borchelt DR. Genetic neurodegenerative disease: the human illness and transgenic model. Science 1998;282: 1079-1082.
- Haass C, Selkoe DJ. Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell 1993;75(6): 1039-1042.
- Ancolio K, Dumanchin C, Barelli H, et al. Unusual phenotypic alter- ation of b amyloid precursor protein (bAPP) maturation by a new Val-715®Met bAPP-770 mutation responsible for probable early-onset Alzheimer’s disease. Proc Natl Acad Sci USA 1999;96:4119-4124.
- Hussain I, Powell D, Howlett DR, et al. Identification of a novel aspar- tic protease (Asp 2) as beta-secretase. Mol Cell Neurosci 1999;14(6): 419-427.
- Yan R, Bienkowski MJ, Shuck ME, et al. Membrane-anchored aspar- tyl protease with Alzheimer’s disease beta-secretase activity. Nature 1999;402(6761):533-537.
- Vassar R, Bennett BD, Babu-Khan S, et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999;286(5440):735-741.
- Sinha S, Anderson JP, Barbour R, et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 1999;402(6761):537-540.
- Saunders AJ, Kim TW, Tanzi RE. BACE homolog, BACE2, reside in the obligate Down syndrome region of chromosome 21. Science 1999;286(5443):1255.
- De Strooper B. Closing in on g-secretase. Nature 2000;405:627-629.
- 37. Li YM, Xu M, Lai MT, et al. Photoactivated g-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 2000;405: 689-693.
- De Strooper B, König G. A firm base for drug development. Nature 1999;402: 471.
- Pericak-Vance MA, Bebout JL, Gaskell PC, et al. Linkage studies in familial Alzheimer disease: evidence for chromosome 19 linkage. Am J Hum Genet 1991;48:1034-1050.
- Strittmatter WJ, Weisgraber KH, Huang DY, et al. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci USA 1993;90:8098-8102.
- Gomez-Isla T, West HL, Rebeck GW, et al. Clinical and pathological correlates of apolipoprotein E epsilon 4 in Alzheimer’s disease. Ann Neurol 1996;39:62-70.
- Schmechel DE, Saunders AM, Strittmatter WJ, et al. Increased amy- loid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA 1993;90:9649-9653.
- Corder EH, Saunders AM, Risch NJ, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet 1994;7:180-184.
- Smith JD, Sikes J, Levin JA. Human apolipoprotein E allele-specific brain expressing transgenic mice. Neurobiol Aging 1998;19:407-413.
- Bales KR, Verina T, Dodel RC, et al. Lack of apolipoprotein E dramati- cally reduces amyloid beta-peptide deposition. Nat Genet 1997;17: 263-264.
- Holtzman DM, Bales KR, Tenkova T, et al. Apolipoprotein E isoform- dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 2000;97: 2892-2897.
- Buttini M, Akeefe H, Lin C, et al. Dominant negative effects of apolipoprotein E4 revealed in transgenic models of neurodegenerative disease. Neuroscience 2000;97:207-210.
- Schenk D, Barbour R, Dunn W, et al. Immunization with amyloid-b attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999;400:173-177.
- Morgan D, Diamond DM, Gottschall PE, et al. A beta peptide vaccina- tion prevents memory loss in an animal model of Alzheimer’s disease. Nature 2000;408(6815):982-985.
- Janus C, Pearson J, McLaurin J, et al. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature 2000;408(6815):979-982.
- Gerlai R. Alzheimer’s disease: beta-amyloid hypothesis strengthened! Trends Neurosci 2001;24:199.
- Chapman PF. Alzheimer’s disease: model behaviour. Nature 2000; 408(6815):915-916.
- Ingram DK. Vaccine development for Alzheimer’s disease: a shot of good news. Trends Neurosci 2001;24:305-307.
- van-Slegtenhorst M, Lewis J, Hutton M. The molecular genetics of the tauopathies. Exp Gerontol 2000;35:461-471.
- Baker M, Kwok JB, Kucera S, et al. Localization of frontotemporal dementia with parkinsonism in an Australian kindred to chromosome 17q21-22. Ann Neurol 1997;42:794-798.
- Nasreddine ZS, Loginov M, Clark LN, et al. From genotype to pheno- type: a clinical pathological, and biochemical investigation of fronto- temporal dementia and parkinsonism (FTDP-17) caused by the P301L tau mutation. Ann Neurol 1999;45:704-715.
- Gotz J, Chen F, Barmettler R, Nitsch RM. Tau filament formation in transgenic mice expressing P301L tau. J Biol Chem 2001;276:529-534.
- Lewis J, McGowan E, Rockwood J, et al. Neurofibrillary tangles, amyo- trophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 2000;25:402-405.
- Hardy J, Duff K, Hardy KG, Perez-Tur J, Hutton M. Genetic dissec- tion of Alzheimer’s disease and related dementias: amyloid and its relationship to tau. Nat Neurosci 1998;1:355-358.
- Gotz J, Chen F, van-Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 2001;293(5534):1491-1495.
- Lewis J, Dickson DW, Lin WL, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001;293(5534):1487-1491.
- Capsoni S, Ugolini G, Comparini A, Ruberti F, Berardi N, Cattaneo A. Alzheimer-like neurodegeneration in aged antinerve growth factor transgenic mice. Proc Natl Acad Sci USA 2000;97:6826-6831.
- Mobley WC, Rutkowski JL, Tennekoon GI, Gemski J, Buchanan K, Johnston MV. Nerve growth factor increases choline acetyltransferase activity in developing basal forebrain neurones. Brain Res 1986;387: 53-62.
- Hefti F. Nerve growth factor promotes survival of septal cholinergic neurones after fimbrial transections. J Neurosci. 1986;6:2155-2162.
- Smith DE, Roberts J, Gage FH, Tuszynski MH. Age-associated neuronal atrophy occurs in the primate brain and is reversible by growth factor gene therapy. Proc Natl Acad Sci USA 1999;96:10893-10898.
- Chen KS, Nishimura MC, Armanini MP, Crowley C, Spencer SD, Phillips HS. Disruption of a single allele of the nerve growth factor gene results in atrophy of basal forebrain cholinergic neurones and memory deficits. J Neurosci. 1997;17:7288-7296.
- Ruberti F, Capsoni S, Comparini A, et al. Phenotypic knockout of nerve growth factor in adult transgenic mice reveals severe deficits in basal forebrain cholinergic neurones, cell death in the spleen, and skeletal muscle dystrophy. J Neurosci. 2000;20:2589-2601.
- Jaffar S, Counts SE, Ma SY, et al. Neuropathology of mice carrying mutant APP(swe) and/or PS1(M146L) transgenes: alterations in the p75(NTR) cholinergic basal forebrain septohippocampal pathway. Exp Neurol 2001;170:227-243.
- Fukuta T, Nitta A, Itoh A, Furukawa S, Nabeshima T. Difference in toxicity of beta-amyloid peptide with aging in relation to nerve growth factor content in rat brain. J Neurol Trans 2001;108: 221-230.
- Rattray M. Is there nicotinic modulation of nerve growth factor? Implications for cholinergic therapies in Alzheimer’s disease. Biol Psychiatry 2001;49:185-193.
- Wolfe MS. Secretase targets for Alzheimer’s disease: identification and therapeutic potential. J Med Chem 2001;44:2039-20-
